Abstract
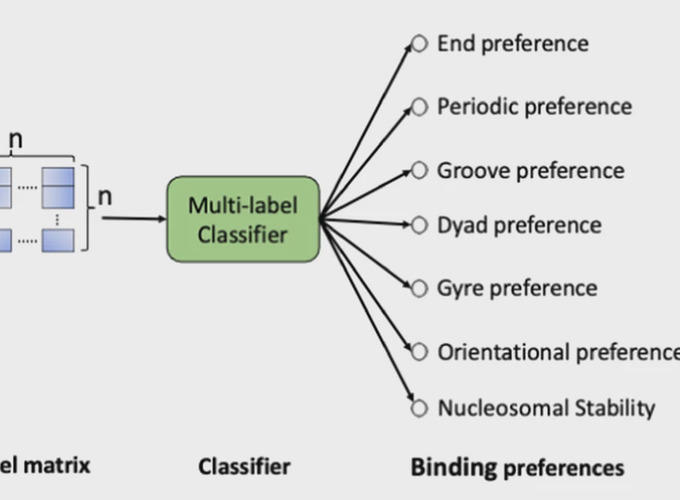
Most transcription factors (TFs) compete with nucleosomes to gain access to their cognate binding sites. Recent studies have identified several TF-nucleosome interaction modes including end binding (EB), oriented binding, periodic binding, dyad binding, groove binding, and gyre spanning. However, there are substantial experimental challenges in measuring nucleosome binding modes for thousands of TFs in different species. Here, we present a computational prediction of the binding modes based on TF protein sequences. With a nested cross-validation procedure, our model outperforms several fine-tuned off-the-shelf machine learning (ML) methods in the multi-label classification task. Our binary classifier for the EB mode performs better than the ML methods with the area under precision-recall curve achieving 75%. The end preference of most TFs is consistent with low nucleosome occupancy around their binding site in GM12878 cells. The nucleosome occupancy data is used as an alternative dataset to confirm the superiority of our EB classifier. This classifier is applied to all TFs in five model organisms, which reveals that the vast majority (88-99%) of TFs have the EB mode, with mammalian TFs having the lowest fraction and yeast TFs having the highest fraction. This work provides the first comprehensive analysis of nucleosome binding modes of TFs.